La nouvelle approche appliquée au secteur des dispositifs médicaux
Organisation
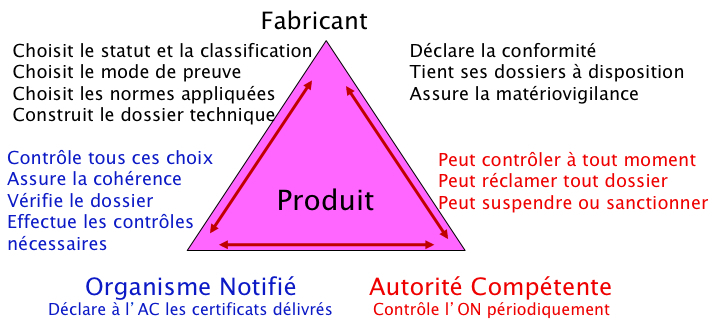
La nouvelle approche repose sur une évaluation décentralisée qui répond aux grands principes suivants :
Elle se fait sous la responsabilité du fabricant. Cette évaluation se fait sur la base de la réponse à des exigences essentielles de sécurité et de performances cliniques qui concernent à la fois la conception des dispositifs mais aussi leur fabrication au fil du temps. Si la réponse aux exigences essentielles applicables est la même pour l'ensemble des DM, la démonstration de la conformité à celles-ci est graduée en fonction de la classe de risque des DM concernés.
Elle nécessite une certification par un tiers habilité : l'organisme notifié (ON). L'ON est désigné (ou « notifié ») par son autorité compétente (AC) nationale (en France, l'Agence nationale de sécurité du médicament et des produits de santé [ANSM]), après une évaluation conjointe de cette autorité, de deux autres AC européennes et de la Commission européenne (CE). Les ON sont notamment soumis à des règles de compétence, d'impartialité et d'indépendance.
Elle impose un contrôle du marché par les autorités sanitaires compétentes (en France l'ANSM) une fois le DM mis sur le marché et cela en sus des audits au moins annuels menés par les ON sur les entreprises. Ainsi en France par exemple, l'ANSM a un rôle de surveillance du marché et l'exerce par ses activités de vigilance, des inspections sur site, des contrôles de produits ou des contrôles réglementaires. Les AC ont, de plus, des pouvoirs dit de police sanitaire permettant de retirer des produits déjà sur le marché, d'interdire, de restreindre ou de suspendre leur mise sur le marché en cas de problématique sanitaire ou de santé publique.
Enfin, tout produit entrant dans le champ de la réglementation doit s'y conformer pour bénéficier de la libre-circulation au sein de l'Union Européenne.
Territoire
Les réglementations de type "nouvelle approche" s'appliquent dans tous les États membres de l'UE et à certains de leurs territoires : Açores, îles Canaries, Guadeloupe, Guyane française, Madère, Martinique, Réunion, Saint-Barthélemy et Saint-Martin.
Par ailleurs, des accords spécifiques existent entre l'UE et certains pays tiers. C'est le cas de l'accord sur l'Espace économique européen (AELE) conclu avec l'Islande, le Liechtenstein et la Norvège. Cet accord étend le marché intérieur à ces trois États communément appelés «États AELE/EEE».
C'est également le cas de l'accord avec la Suisse et de l'accord d'union douanière conclu entre l'UE et la Turquie visant à garantir la libre circulation des produits entre l'UE et la Turquie, en supprimant à la frontière UE-Turquie les contrôles à l'importation de ces produits.
Gouvernance
La gouvernance, c'est-à-dire l'ensemble des structures mises en place pour régir le secteur des dispositifs médicaux, est essentiellement européenne.
Au niveau européen
Il existe plusieurs groupes de travail de la Commission dans le champs des dispositifs médicaux :
Medical Devices Expert Group (MDEG)
Competent Authority (CA) meetings
Vigilance
Classification and Borderline
Compliance and Enforcement Group (COEN)
Notified Body Operations Group (NBOG)
IVD Technical Group
Working Group on Clinical Investigation and Evaluation (CIE)
Electronic Labelling Working Group
New & Emerging Technologies Working Group (NET)
Eudamed Working Group
NB-MED
La liste des groupes est accessible sur le site de la commission.
Medical Devices Expert Group (MDEG)
Le Medical Devices Expert Group (MDEG) est le groupe principal de la Commission européenne dans le secteur des dispositifs médicaux.
Plusieurs publications émanent du MDEG ou de « sous-groupe du MDEG »
Exemples :
Guidance MEDDEVs (cf module spécifique)
- guidances notes (fabricant de DM de classe I, sur mesure ...)
- guidance documents (reclassification 2005/50/CE)...
- own brand labellers,
- rapport avec la Directive 89/686/CEE sur les équipements de protection individuelle...
Remarque : Évolution avec le nouveau règlement européen
Le règlement 2017/745 établit une nouvelle gouvernance et crée le GCDM (Groupe de coordination en matière de dispositifs médicaux).
Remarque : Groupe de coordination en matière de dispositifs médicaux (GCDM)
La composition, les objectifs et les missions du Groupe de coordination en matière de dispositifs médicaux (GCDM) sont définis dans les articles 103 à 105 du règlement. Le GCDM sera organisé en 6 clusters qui réuniront chacun un ou des sous-groupes de travail :
- Pré-market,
- Post-market and clinical,
- New technologies,
- Systems (Eudamed, UDI),
- International Matters,
- IVD
Le GCDM, travaillera en collaboration avec le groupe de coordination des ON (article 49) et le comité d'expert des dispositifs médicaux (article 114).