Modalités d'évaluation de la conformité
Contexte
Comme indiqué dans le module « principes de l'évaluation de la conformité », le fabricant choisit la voie de démonstration réglementaire. Pour un dispositif médical, les différentes combinaisons d'annexes sont décrites dans l'article 11 de la Directive Dispositifs Médicaux (93/42/CEE).
Référentiels d'évaluation de la conformité : les annexes des directives
Les annexes choisies pour établir la conformité sont mentionnées sur le certificat CE.
Le diagramme ci-après illustre les différentes combinaisons possible pour une classe de dispositif afin d'obtenir le marquage CE d'un dispositif médical.
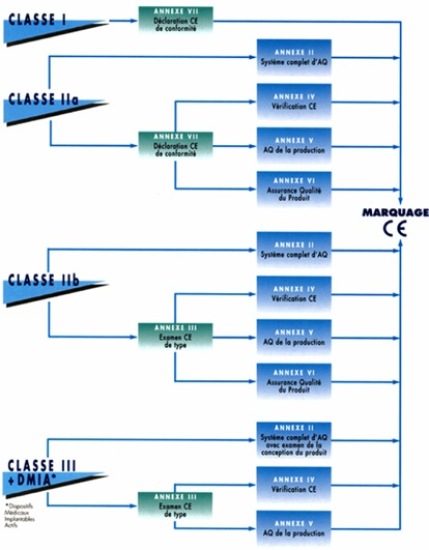
Annexe II : Système complet d'Assurance Qualité :
L’organisme notifié audite de système qualité du fabricant. La norme européenne harmonisée 13 485 est la norme de référence concernant les systèmes de management de la qualité pour la conception, la fabrication, le contrôle final et les essais des dispositifs médicaux
Annexe III : Examen CE de type
L'examen CE de type, délivré par l'Organisme Notifié, porte sur la conception et doit être complété par un module prévoyant l'évaluation en phase de production.
Annexe IV : Vérification CE
Elle porte sur la phase de production et complète l'annexe III. L'Organisme Notifié contrôle la conformité au « type » décrit dans l'attestation d'examen CE de type et délivre un certificat de conformité.
Annexe V : Assurance qualité de la production
L'Organisme Notifié contrôle et approuve le système qualité mis en place pour la production, le contrôle final et les essais.
Annexe VI : Assurance Qualité des Produits
L'Organisme Notifié contrôle et approuve le système qualité mis en place pour le contrôle final et les essais.
Annexe VII : Déclaration CE de conformité
Le fabricant déclare, dans une déclaration de conformité, que son dispositif répond aux exigences essentielles de la directive.
Situation particulière : dispositif de classe I stérile et I de mesurage.
Dans ce cas, le fabricant doit établir une déclaration de conformité et faire auditer par un organisme notifié, selon les annexes II, IV, V ou VI :
— dans le cas des produits mis sur le marché à l'état stérile, aux seuls aspects de la fabrication liés à l'obtention et au maintien de l'état stérile,
— dans le cas des dispositifs ayant une fonction de mesurage, aux seuls aspects de la fabrication liés à la conformité des produits aux exigences métrologiques.
Modalités d'évaluation de la conformité par l'organisme notifié
Après une première prise de contact et l'étude de la recevabilité de la demande par l'ON (le produit est bien un DM correspondant au champ de désignation de l'ON, l'entreprise a une existence juridique et ses différents sites sont identifiés...) le processus de certification est déroulé de la façon suivante :
évaluation : audit(s) du système de management de la qualité sur site du fabricant et de ses sous-traitants critiques d'une part et évaluation(s) de la documentation technique (dont démonstration clinique) d'autre part. Les modalités précises de ces évaluations en matière de durée, périodicité, sites à auditer, profondeur de l'analyse, systématisation de l'évaluation complète de la documentation technique ou échantillonnage dans le temps... dépendent de la taille de l'entreprise, du nombre et des catégories des produits, de leur classe de risque, de l'existence de sous-traitants critiques, de la procédure sélectionnée... ;
prise en compte des plans d'actions du fabricant au regard des éventuels écarts relevés lors de ces évaluations ;
revue indépendante des résultats finaux de ces évaluations pour contrôler la démonstration de conformité mise en œuvre par le fabricant ;
décision de certification et émission des certificats correspondants pour une durée maximale de 5 ans.