Acteurs de l'évaluation de conformité
Le fabricant, l'organisme notifié et l'autorité compétente
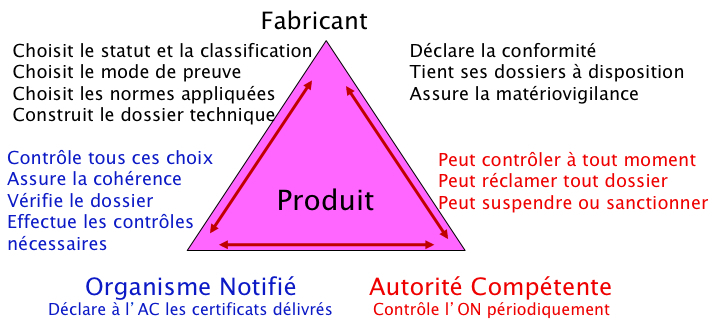
Le fabricant
Le fabricant, responsable du dispositif médical doit tout d'abord vérifier que le produit qu'il développe répond bien à la définition du dispositif médical, et appliquer les règles de classification de l'annexe IX. Si le fabricant est établi en dehors du territoire de l'Union Européenne, il doit désigner un mandataire dont le siège social est situé dans un des pays communautaires.
Si le produit est un dispositif médical de classe I (non stérile et sans fonction de mesurage), le fabricant auto-certifie ses procédures de conformité. L'autorité compétente pourra procéder ultérieurement à un contrôle de conformité dans le cadre de ses activités de surveillance du marché.
Si le dispositif relève d'une autre classe, le fabricant devra choisir librement un organisme notifié parmi ceux désignés par les autorités compétentes de l'UE, dont le numéro d'identification apparaîtra aux côtés du symbole CE sur le dispositif une fois mis sur le marché. En cas de litige d'interprétation sur le statut ou la classification, l'autorité compétente dont relève l'organisme notifié est saisie pour arbitrage.
Le fabricant doit rédiger la documentation technique démontrant la conformité du produit aux exigences essentielles de l'annexe I de la directive concernée, quelle que soit la classe du dispositif médical. Pour ce faire, il devra soit se conformer aux normes harmonisées existantes, soit justifier son choix d'un autre référentiel, soit, s'il s'agit d'un dispositif innovant (ou en cas d'absence de norme harmonisée), décrire les modalités choisies pour la démonstration de conformité aux exigences essentielles.
Il devra alors se soumettre à l'une des procédures de certification prévues par la directive pour la classe considérée. Dans le cas d'un dispositif incorporant un médicament, le fabricant doit également consulter une autorité compétente en matière de médicament.
Après obtention du certificat délivré par l'organisme notifié, d'une durée de validité maximale de cinq ans, le fabricant pourra déclarer la conformité, apposer le marquage CE (décrit à l'annexe XII) et mettre sur le marché le dispositif médical dans l'ensemble des pays de l'Union européenne. ajouter le lien vers le chapitre "nvelle approche DM - territoire"
Durant cette période :
le fabricant doit notamment mettre en place un système de surveillance après commercialisation. L'ensemble des informations collectées peut conduire à des mesures correctives et des améliorations incrémentales des dispositifs ;
le fabricant est également tenu de signaler les incidents ou risques d'incidents graves à l'autorité compétente du pays concerné, de les analyser et prendre les mesures nécessaires pour éviter la survenue de tout nouvel incident, comme par exemple, les retraits qui doivent, par ailleurs, être signalés à l'autorité compétente ;
l'organisme notifié effectue régulièrement des audits de suivi du système de qualité ainsi que du respect des règles établies pour la surveillance post-commercialisation.
Les directives spécifiques au DM confient à chaque autorité compétente sur son territoire national (en France, l'ANSM) trois missions principales relatives à la désignation et à la surveillance des organismes notifiés, à la surveillance du marché et à la vigilance (lien vers la partie concernant l'ANSM).
Organisme notifié
Les organismes notifiés (ONs) sont des organisations qui sont soumises à des règles précises d'habilitation et d'exercice. Ce sont les autorités compétentes (en France, l'ANSM) qui habilitent, surveillent et renouvellent ces organismes dans un processus impliquant la Commission Européenne et d'autres autorités compétentes sur la base de plusieurs critères : compétences, impartialité et indépendance.
Les ON sont désignés en fonction de leurs compétences sur tout ou partie du champ d'application de la réglementation. Si le choix de l'ON est libre, les règles de certification sont fixées par la réglementation et sont les mêmes quel que soit l'ON.
Les organismes notifiés évaluent systématiquement, de manière indépendante, la documentation technique du DM et auditent le système de management de la qualité des différents sites du farbicant ainsi que ceux de ses sous-traitants critiques. Si ces évaluations satisfont aux exigences, l'organisme notifié leur délivre alors un certificat de marquage CE médical valide pour une durée limitée à 5 ans au maximum.
Autorité compétente (en France, ANSM)
Les autorités compétentes (en France l'ANSM) ont un rôle de surveillance du marché et l'exerce par leurs activités de vigilance, d'inspections sur site, de contrôles de produits ou de contrôles réglementaires. Les AC ont, de plus, des pouvoirs dit de police sanitaire permettant de retirer des produits déjà sur le marché, d'interdire, de restreindre ou de suspendre leur mise sur le marché en cas de problématique sanitaire ou de santé publique.
Ils sont par ailleurs responsables de l'habilitation et de la surveillance des ON.
Texte légal : Organismes notifiés
Deux textes portent sur le fonctionnement des organismes notifiés :
Règlement d'exécution (UE) n° 920/2013 de la Commission du 24 septembre 2013 relatif à la désignation et au contrôle des organismes notifiés au titre de la directive 90/385/CEE du Conseil concernant les dispositifs médicaux implantables actifs et de la directive 93/42/CEE du Conseil relative aux dispositifs médicaux (JOUE 25-9-2013)
Recommandation de la Commission du 24 septembre 2013 relative aux audits et évaluations réalisés par les organismes notifiés dans le domaine des dispositifs médicaux (JOUE 25-9-2013)
Le premier texte précise les critères auxquels doivent satisfaire les organismes notifiés qui sont chargés d'inspecter les fabricants de dispositifs médicaux. Le second précise les missions incombant à ces organismes à l'occasion des audits et des évaluations qu'ils effectuent dans le secteur des dispositifs médicaux